Understanding genetic processes in cultivated plants must include both qualitative and quantitative traits. Many important characteristics as for example petal number, flower size, the amounts of secondary metabolites involved in flower colour and scent as well as disease resistance are in part determined by quantitative genetic factors. Knowledge gained in model plants does not completely explain genetic processes in crops as genome projects conducted over the last decade uncovered numerous differences between plant species. Therefore, detailed analyses of genetic processes need to be conducted directly in individual crop plants and not only in model plants.



Analysis of the rose genome
As partners of an international consortium we contributed to the generation of a high quality chromosome scale rose genome sequence. The chromosome scale assembly comprises 512 Mb of the predicted genome size of 533 Mb. The 44481 genes comprise 39669 protein-coding genes of which 6543 are specific to the genus Rosa and 5,867 proteins which have no homologue in Arabidopsis thaliana. The high quality of the assembly now allows to analyse genetic processes based on structural/positional information with an unprecedented precision.
https://www.rosaceae.org/species/rosa/chinensis/genome_v1.0 GDR website with the rose genome information
https://www.nature.com/articles/s41477-018-0166-1 Publication of the rose genome in Nature Plants
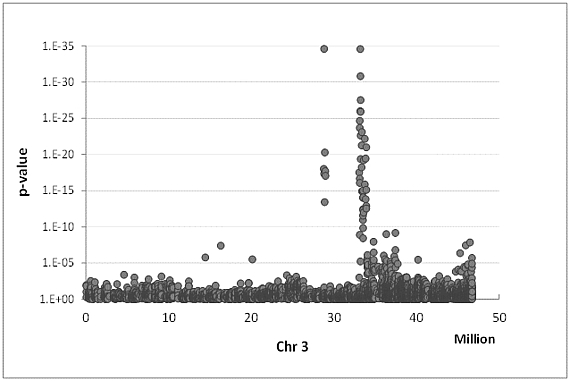
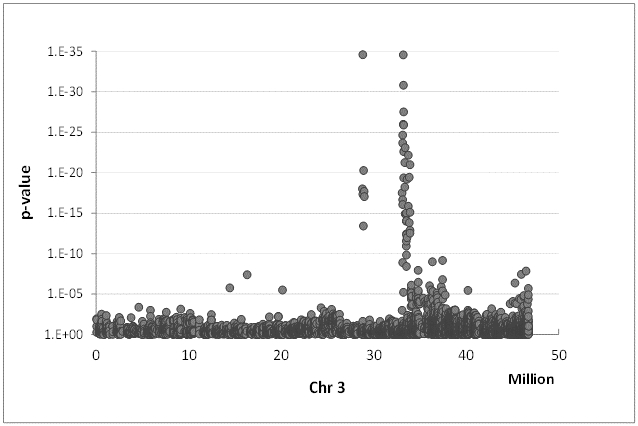
High throughput genotyping in roses
In contrast to major crop species (like maize, rice or potatoes) breeding and selection using molecular marker information is still in its infancy in ornamentals. Mainly PCR based markers are used in relative low numbers of up to only few hundreds. To achieve a dense coverage of the seven rose chromosomes with molecular markers, scientist from the Wageningen UR Plant Breeding in the Netherlands and from the Molecular Plant Breeding Department in Hannover developed the WagRhSNP Axiom array containing 68.893 SNPs (single nucleotide polymorphisms). The commercially available chip can now be used by rose researchers to identify candidate genes for many different traits with very high precision. In addition, a rose transcriptome database for more than 13.000 full length genes was developed.
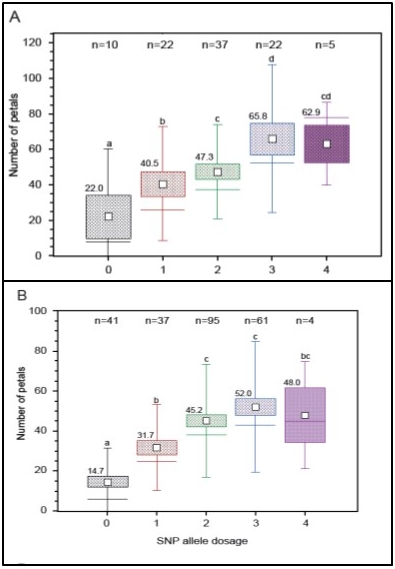
Associations between traits and markers
Most of the phenotypic traits in living organisms are regulated by more than a single gene. The detection of the responsible loci can be done in progenies from a cross of two genotypes by QTL (quantitative trait loci) analysis or in much higher genetic resolution by genome wide association studies (GWAS) in populations of unrelated genotypes. Beside several QTL studies we also established a GWAS population of 96 mostly tetraploid rose genotypes which is phenotyped for 20 different traits and genotyped using the WagRhSNP array with around 68,000 SNPs, AFLPs and SSRs. For nearly all traits we could identify closely linked markers having a significant effect on the phenotype. Some of which were already validated in around 240 rose genotypes from a collection of the Bundessortenamt in Hannover and can be used for marker assisted selection in roses.





